Sindrome di Brugada
Cos’è la Sindrome di Brugada?
La sindrome di Brugada è una sindrome aritmica, geneticamente determinata che predispone allo sviluppo di disturbi del ritmo cardiaco anche gravi, che possono provocare morte improvvisa.
Gli eventi aritmici della malattia si presentano solitamente di notte, caratteristica per la quale questa patologia viene denominata con nome diverso in varie regioni asiatiche: “Lai Tai” (morte nel sonno) in Thailandia, “Bangungut” (urlo seguito da morte improvvisa durante il sonno) nelle Filippine, “Pokkuri” (morte improvvisa ed inaspettata durante la notte) in Giappone.
La sindrome di Brugada deve il suo nome a due fratelli cardiologi spagnoli, Pedro e Josep Brugada che la descrissero compiutamente nel 1992.
Tuttavia già negli anni ’80 i cardiologi italiani Nava, Martini e Thiene ne delinearono le caratteristiche principali sul Giornale Italiano di Cardiologia.


Quanto è frequente la Sindrome di Brugada?
La sindrome di Brugada è una condizione rara: secondo stime attendibili infatti, ne sarebbero affetti da 1 a 30 persone ogni 10.000 individui considerati
La massima diffusione si manifesta tra le popolazioni di origine Asiatica, in particolari tra le persone originarie di Thailandia e Laos. Secondo quanto riporta la letteratura medica a riguardo, la Sindrome di Brugada sarebbe la causa del 4-12% di tutti i casi di morte cardiaca improvvisa e di più del 20% di tutti i casi di morte cardiaca improvvisa in persone con un cuore strutturalmente sano.
Quali sono le cause che determinano la Sindrome di Brugada?
La Sindrome di Brugada è una malattia determinata da alterazioni genetiche e si trasmette da una generazione all’altra con modalità cosidetta autosomica dominante.
Ciò significa che è sufficiente che un solo genitore presenti una anomalia di un gene alterato perché possa trasmettere la malattia alla prole.
In circa il 40% dei casi l’anomalia genetica può essere determinata con il riscontro di una specifica alterazione di un gene. Ad oggi sono note 18 diverse anomalie genetiche che possono causare la malattia. Tali anomalie producono un difetto elettrico dei canali ionici che determinano la conduzione dell’impulso sulle membrane delle cellule cardiache. Da questo difetto l’attività elettrica del cuore può subire improvvise alterazioni che determinano aritmie talora anche mortali. Il gene più frequentemente coinvolto è quello SCNA5A che codifica per una subunità di un canale che consente il passaggio di ioni sodio attraverso la membrana della cellula cardiaca.
Nel 60% dei casi non siamo in grado a tutt’oggi di determinare quale sia il preciso meccanismo che determina la malattia sia esso genetico o di altra natura.


Come si manifesta la Sindrome di Brugada?
La maggior parte dei pazienti affetti da sindrome di Brugada rimane asintomatico (70%).
Nel 30% dei casi, circa, i pazienti manifestano dei sintomi di gravità variabile che si presentano più frequentemente in giovani maschi (con rapporto maschi/femmine di 8:1) con età compresa tra 30 e 40 anni. In letteratura sono tuttavia descritti casi in un range di età molto ampio (6-77 anni).
I sintomi possono essere batticuore, offuscamento della vista, vertigini improvvise, senso di mancamento, senso di oppressione toracica.
Sintomo cardine nei pazienti con Sindrome di Brugada è la perdita di coscienza improvvisa o sincope. La sincope aritmica è caratteristicamente subitanea, non preceduta da sintomi premonitori a differenza di quella non aritmica cosiddetta vagale o neuromediata che si manifesta dopo un corteo di sintomi premonitori, anch’essa frequente nei pazienti con Brugada.
La morte improvvisa, pur rara rappresenta, nel 5% dei casi il primo (ed ultimo) sintomo della malattia senza alcun segno premonitore.
I sintomi non si verificano quasi mai sotto sforzo, nella maggior parte dei casi invece a riposo, dopo sforzo, durante la digestione, di notte poiché il rallentamento della frequenza cardiaca e l’aumentato tono vagale sono fattori favorenti la genesi delle aritmie che stanno alla base della sintomatologia.
Nei casi più drammatici si può verificare la cosiddetta tempesta aritmica che consiste nel susseguirsi di aritmie ventricolari che portano a svenimenti o veri e propri arresti cardiaci subentranti che obbligano ad interventi urgenti di rianimazione e di stabilizzazione elettrica.
Come si diagnostica la Sindrome di Brugada?
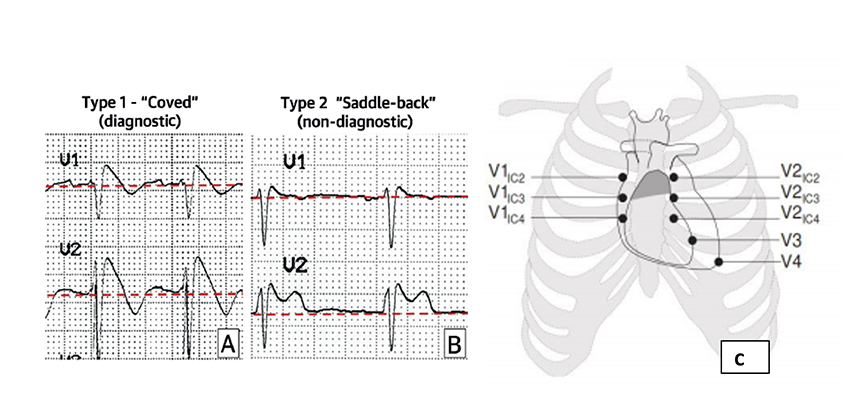
La diagnosi della sindrome di Brugada si fa con l’elettrocardiogramma. I pazienti affetti presentano alterazioni che sono localizzate in una ristretta area del tessuto cardiaco situata sulla superficie esterna dell’infundibolo del ventricolo destro ad di sotto dell’epicardio. Gli effetti di questa alterazione possono’ essere registrati dall’elettrocardiogramma, in particolare dagli elettrodi che si posizionano nel II°-III°-IV° spazio intercostale, proprio al di sopra dell’area malata (fig 1).
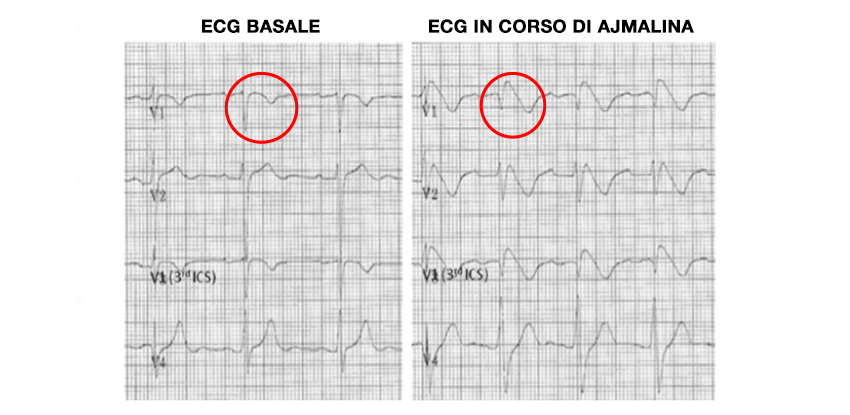
Quando la alterazione è conclamata si parla di pattern Brugada TIPO 1 : la registrazione di questo pattern consente di fare la diagnosi certa della sindrome. La registrazione invece di pattern tipo 2 e tipo 3 pone solo il sospetto della presenza della sindrome che deve essere confermata mediante test provocativi: test all’ajmalina (infusione di ajmalina o flecainide) che fanno emergere il pattern TIPO 1 nei soggetti affetti ( fig 2) Infatti il pattern TIPO 1 puo’ essere intermittente: quando è presente indica una corrente elettrica anomala attiva che puo’ dare luogo ai fenomeni di cortocircuito che innescano le aritmie.
Per effettuare DIAGNOSI CERTA è necessario registrare il pattern TIPO 1( fig. 1) spontaneamente se presente o facendolo emergere con test provocativi.

Come si valuta il rischio aritmico nella Sindrome di Brugada?
Il rischio di sviluppare eventi aritmici nei pazienti affetti e’ molto variabile.
La presenza di batticuore improvviso, una sensazione di offuscamento della vista, vertigini costituiscono segnale di interesse da parte del cardiologo in quanto possibili marker di brevi aritmie e quindi di rischio.
La familiarità per morte improvvisa giovanile in parenti di primo grado aumenta il rischio aritmico in maniera significativa.
Mentre dati meno certi esistono per parenti di grado più elevato o per eventi in età avanzata.
La sincope (svenimento a ciel sereno), la registrazione di aritmie ventricolari maggiori complesse e infine l’arresto cardiaco abortito sono considerati degli importanti fattori di rischio per la morte improvvisa. Nei pazienti con episodi di sincope bisogna escludere gli eventi legati a fenomeni benigni di abbassamento di pressione e frequenza legati a riflessi vagali (sincope neuromediata) che non hanno impatto prognostico significativo. I pazienti invece con episodi di sincope a ciel sereno manifestano un rischio di eventi gravi annui di circa il 2%.
I pazienti sopravvissuti ad arresto cardiaco hanno 8% di rischio annuo di recidiva costituiscono il sottogruppo a rischio più elevato.
La completa assenza di sintomi costituisce un dato rassicurante, anche se talora gli eventi avversi gravi possono verificarsi in soggetti che non hanno mai manifestato segni premonitori. Questo gruppo di pazienti rappresenta la maggiore sfida di valutazione del rischio da parte del cardiologo.

La presenza all’ECG del pattern tipo 1 puo’ essere di aiuto in questa valutazione: se presente spontaneamente senza necessita’ di indurlo con test provocativi, il rischio aritmico e’ piu’ elevato poiche’ indica la presenza attiva della corrente elettrica anomala. Questo sottogruppo di pazienti presenta un rischio di morte improvvisa di dell’1-2% all’anno. E’ necessario quindi ricercare l’emergere spontaneo del pattern TIPO 1 con una registrazione holter a 12 derivazioni nell’arco delle 24 h e non limitarsi alla registrazione di un singolo elettrocardiogramma.
Se il pattern Tipo 1 non si manifesta mai spontaneamente il rischio aritmico è basso. Questo dato rassicurante va comunque confermato con holter ripetuti nel tempo per escludere una evoluzione sfavorevole del rischio.
Sempre in pazienti a rischio intermedio la stratificazione del rischio aritmico puo’ valersi anche dello studio elettrofisiologico. Questo esame consiste nella stimolazione del ventricolo destro (definita programmata) mediante impulsi in grado di evocare risposte aritmiche anomale in pazienti con particolare vulnerabilita’ elettrica. Tale vulnerabilita’ se presente costituisce un ulteriore importante indicatore di rischio aritmico in pazienti affetti da Sindrome di Brugada.
Il clinico inoltre si vale, nella valutazione complessiva del rischio di altri elementi come la concomitante presenza di fibrillazione atriale, di malattia del nodo del seno, di presenza di complessi QRS allargati, frammentati all’ECG, di ripolarizzazione precoce, blocco atrioventricolare.
Sulla base di tutti gli elementi sopra esposti il cardiologo formula una valutazione del rischio individualizzata sul singolo paziente e si orienta verso la forma più appropriata di trattamento.